

EU MDR and IVDR Medical Device Regulatory Pathway

How to Register a Medical Device in the European Union (EU): Step-by-Step Guide
The European Union (EU) is one of the world's largest markets for medical technology. To ensure that only safe, effective, and high-quality devices reach patients, manufacturers must comply with the Medical Device Regulation (MDR, EU 2017/745) and the In Vitro Diagnostic Regulation (IVDR, EU 2017/746).
Unlike the U.S. FDA, the EU does not have a single central regulatory authority for medical devices. Instead, compliance is assessed through a multi-level framework involving manufacturers, Notified Bodies (NBs), and National Competent Authorities (NCAs) in each Member State.
This blog provides a step-by-step overview of the EU regulatory pathway for both general medical devices and in vitro diagnostic (IVD) devices.
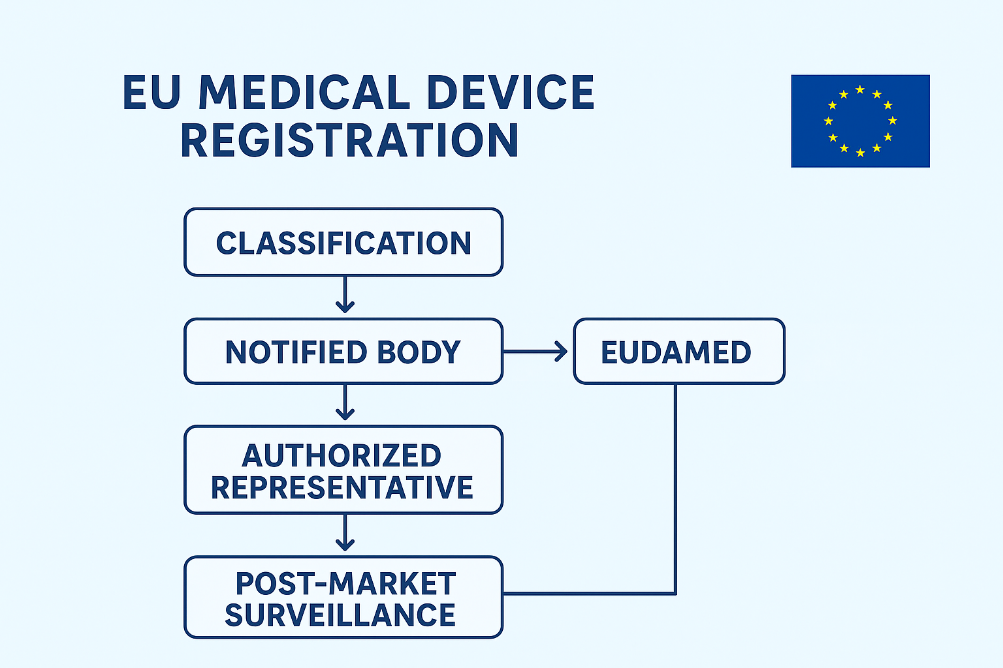
1. Understand the Regulatory Bodies
- There is no single central EU regulatory authority.
- Notified Bodies (NBs): Independent, EU-designated organizations that carry out conformity assessments for medium- and high-risk devices.
Full list: EU Notified Bodies - National Competent Authorities (NCAs): Regulatory agencies in each Member State responsible for market surveillance and enforcement.
List: National Competent Authorities
2. Classify Your Device
Device classification determines the regulatory pathway and level of scrutiny. Medical Devices (MDR - Annex VIII):
- Class I: Lowest risk
- Class IIa: Medium risk
- Class IIb: Medium-high risk
- Class III: Highest risk In Vitro Diagnostic Devices (IVDR - Annex VIII):
- Class A: Lowest risk
- Class B: Moderate risk
- Class C: High risk
- Class D: Highest risk
3. Choose the Correct Regulatory Pathway
Conformity assessment procedures are risk-based:
EU MDR (Article 52):
- Annex IX: Quality management system (QMS) + technical documentation assessment (most common for higher-risk devices).
- Annex X: Type-examination (used often with Annex XI).
- Annex XI: Product conformity verification.
EU IVDR (Article 48):
- Annex IX, X, XI also apply, with adaptations for IVDs.
4. Implement a Quality Management System (QMS)
A robust QMS is mandatory under both MDR and IVDR. It should cover the entire product lifecycle — from design and manufacturing to post-market monitoring. The framework most companies use is ISO 13485:2016, as it aligns closely with EU expectations.
MDR Article 10(9) and IVDR Article 10(8) require that the QMS is proportionate to device class and risk.
5. Compile Technical Documentation
The Technical File (Annex II & III of MDR/IVDR) is the foundation for CE marking. It must be detailed enough for Competent Authorities or Notified Bodies to assess compliance.
Key contents include:
- Device description, variants, and intended purpose
- Design and manufacturing processes
- Risk management documentation
- Verification and validation reports (e.g., biocompatibility, software, sterilization)
- Clinical Evaluation Report (CER) or Performance Evaluation Report (PER)
- Labeling, packaging, IFU, and UDI information
- PMS and PMCF/PMPF plans
Keep the file up to date and searchable, as it will be reviewed during audits and regulatory inspections.
6. Appoint an Authorized Representative (AR)
Non-EU manufacturers must appoint a single Authorized Representative (AR) located in the EU (MDR/IVDR Article 11). The AR's responsibilities include:
- Acting as the liaison with Competent Authorities
- Holding copies of the Technical Documentation and DoC
- Ensuring the manufacturer meets EU obligations
- Cooperating in vigilance and incident investigations The AR shares legal responsibility for compliance, so a formal written agreement is required.
7. Register in EUDAMED
EUDAMED is the EU database for medical devices.
Manufacturers, ARs, and importers must:
- Register as an Actor to obtain a Single Registration Number (SRN)
- Upload device information, including Basic UDI-DI
- Update data when changes occur
Currently, actor and UDI/device registration modules are live; vigilance and surveillance modules are being rolled out.
8. Affix the CE Mark
- Once conformity is demonstrated, issue an EU Declaration of Conformity (DoC) and affix the CE marking.
- For devices assessed by a Notified Body, the CE mark must include the NB's identification number.
- CE mark requirements: Minimum vertical dimension of 5 mm (exceptions for small devices).
- Legal references: Article 20 (MDR) and Article 18 (IVDR).
9. Conduct Post-Market Surveillance (PMS)
Manufacturers must operate a PMS system proportional to the risk class.
- MDR Chapter VII (Articles 83-86) & Annex III
- IVDR Chapter VII (Articles 78-81) & Annex III
PMS includes:
- Post-Market Surveillance Reports (PMSR) for Class I devices
- Periodic Safety Update Reports (PSUR) for Class IIa, IIb, and III devices
- Post-Market Clinical Follow-up (PMCF) or Post-Market Performance Follow-up (PMPF)
10. Unique Device Identification (UDI)
- UDI provides a harmonized identification system across the EU for device traceability and safety monitoring.
- Legal basis:
- MDR Articles 27-28, Annex VI (Part C).
- IVDR Articles 24-25, Annex VI (Part C).
- Application:
- UDI must appear on device labels, packaging, and in some cases directly on the device.
- Manufacturers must upload UDI data into the EUDAMED UDI/Device Registration module.
11. Guidance and Resources
The European Commission provides ongoing guidance via the MDCG (Medical Device Coordination Group):👉 MDCG guidance documents
Conclusion
- The EU regulatory pathway is risk-based and highly structured. To place a device on the EU market, manufacturers must:
- Identify applicable regulation (MDR or IVDR).
- Classify the device correctly.
- Establish a compliant QMS.
- Prepare robust technical documentation.
- Engage with Notified Bodies (if required).
- Register in EUDAMED and implement UDI.
- Affix the CE mark.
- Maintain PMS, vigilance, and continuous compliance.
By following these steps, manufacturers can ensure smooth CE marking and secure access to the European market.
Share this blog
Read More Blogs
Medical Device Approval Process in Japan | PMDA and MHLW Guide

SAHPRA Medical Device Registration: Step-by-Step Guide

FDA's Latest Medical Device Cybersecurity Guidance