

Medical Device Approval Process in Japan | PMDA and MHLW Guide

Japan Medical Device Regulations: Approval Process, PMDA & MHLW Guide
Japan is one of the largest and most technologically advanced medical device markets in the world. However, entering this market requires navigating a rigorous regulatory framework designed to ensure that all devices are safe, effective, and of high quality before reaching patients. Understanding the Japan medical device classification system, submission requirements, and foreign manufacturer obligations is essential for successful market entry.
Regulatory Authorities in Japan
Two main authorities oversee medical device regulation:
- Ministry of Health, Labour and Welfare (MHLW): Responsible for administrative decisions, including product approvals, guidance issuance, and determining whether a product qualifies as a medical device under the Pharmaceuticals and Medical Devices (PMD) Act.
- Pharmaceuticals and Medical Devices Agency (PMDA): Conducts scientific reviews, evaluates safety and efficacy, manages consultations, and monitors post-market safety.
Both agencies work closely to ensure that devices meet Japan’s stringent safety and performance requirements.
Device Classification
- Class I General Medical Devices (Low Risk): Require only notification (Todokede) to PMDA.
- Class II Controlled Medical Devices (Low Risk):
- If certification standards exist → Certification by a Registered Certification Body (RCB).
- If no standards exist → MHLW approval after PMDA review.
- Class III Specially Controlled Medical Devices (Medium Risk):
- With standards → RCB certification.
- Without standards → Approval by MHLW after PMDA review.
- Class IV Specially Controlled Medical Devices (High Risk): Always require MHLW approval after full PMDA review.
Regardless of classification, the Marketing Authorization Holder (MAH) is responsible for ensuring device safety, efficacy, and quality before submission.
Foreign Manufacturer Registration (FMR)
Foreign manufacturers cannot directly submit device applications in Japan. They must first complete Foreign Manufacturer Registration (FMR) with the PMDA and appoint a Japanese MAH or Designated MAH (DMAH). Required FMR documents include:
- Application forms (Form No.18, Form No.16-(2), etc.)
- Curriculum vitae of the responsible person for the facility
- List of products to be exported to Japan
- Manufacturing process details (assembly, sterilization, packaging)
- Facility layouts and floor plans
- License/permit from home country authorities (if applicable)
Registration is typically valid for three years or more and must be renewed periodically.
Pre-Market Submission Pathways
Once a foreign manufacturer completes Foreign Manufacturer Registration (FMR) and appoints a Marketing Authorization Holder (MAH/DMAH), the next step is choosing the correct submission pathway. In Japan, there are three main regulatory routes depending on the medical device classification: Todokede (Notification), Ninsho (Certification), and Shonin (Approval).
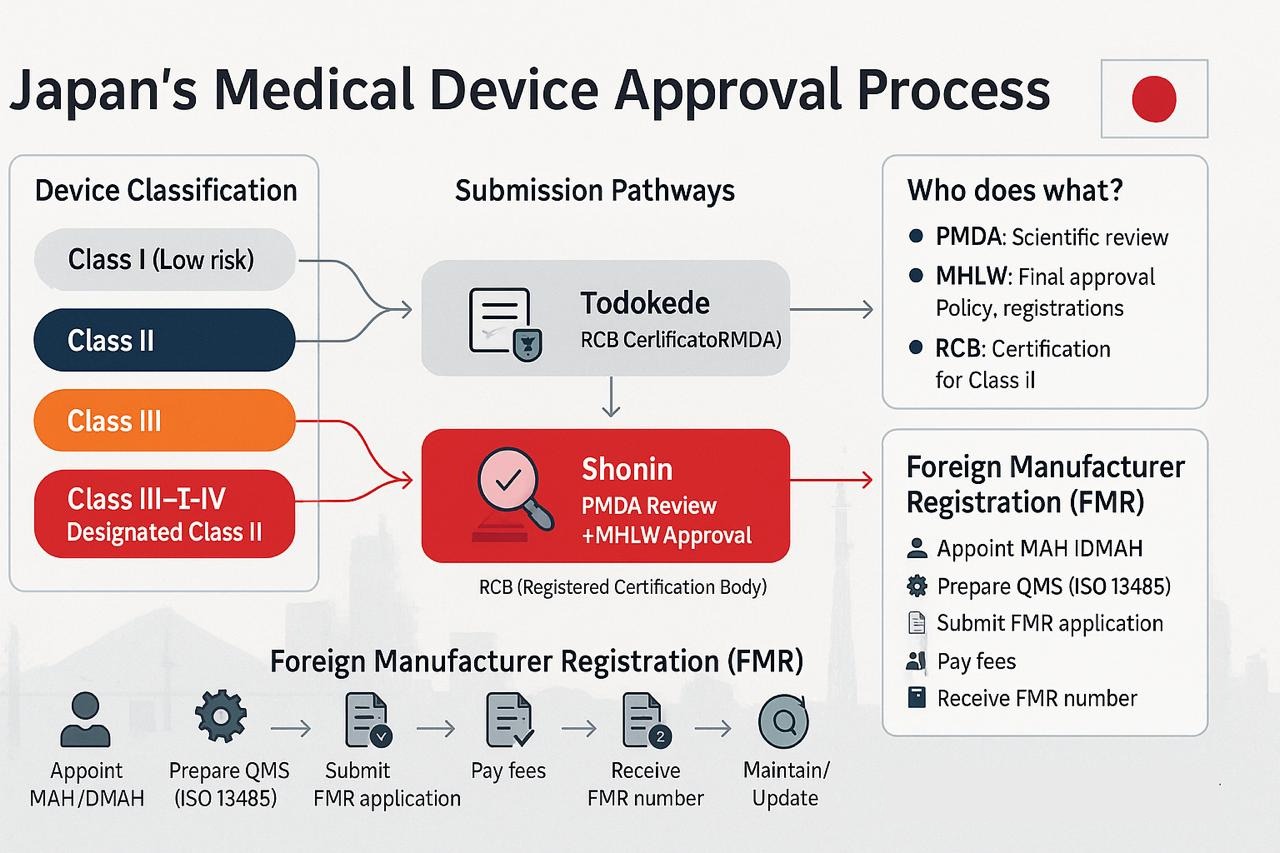
Once FMR is complete, the submission pathway depends on the device class:
Todokede (Notification) - Class I Devices
- Basic device information (JMDN code, intended use, specifications)
- Labelling samples (in Japanese)
- MAH/DMAH information
Ninsho (Certification) - Class II & Some Class III Devices
- Application form
- Device description (technical file)
- Risk classification & JMDN code
- Proof of conformity to Japanese standards (JIS)
- Quality Management System (QMS) conformity documents (audit certificates, QMS report)
- Labelling and Instructions for Use (Japanese)
Shonin (Approval) – Most Class III & All Class IV Devices
- Application dossier to PMDA including:
- Device Master File / STED (Summary of Technical Documentation)
- Clinical data (if required for safety/performance)
- Non-clinical test reports (biocompatibility, electrical safety, software validation, sterilization)
- Manufacturing process details & quality control documentation
- QMS audit results (Ordinance No. 169 compliance)
- Risk analysis documents
- Labeling, packaging, and IFU in Japanese
- Comparison with predicate devices (if applicable)
Typical Review Timelines
- Generic medical device (new product): ~6.8 months
- Generic device (partially modified): ~5.8 months
- Device without clinical trials: ~10.2 months
- Device with clinical trials: ~13.6 months
Quality Management System (QMS)
For Class II–IV devices, compliance with Japan’s QMS (Ordinance No. 169) is mandatory. While ISO 13485 and MDSAP (Medical Device Single Audit Program) certificates support compliance, they do not replace Japanese QMS audits. QMS documentation forms a critical part of the submission dossier.
Post-Market Obligations
After approval or certification, the MAH (Marketing Authorization Holder) must ensure ongoing safety and compliance through:
- Safety reporting: Submission of adverse events from healthcare professionals and patients (Article 68-10, PMD Act)
- Labelling: All labelling and IFU must be in Japanese
- Registration renewals: Periodic renewal of approvals and certifications
- Vigilance: Continuous monitoring of device safety, quality, and performance
Conclusion
Japan's medical device regulatory pathway is highly structured and risk-based. Foreign manufacturers must carefully plan their market entry by:
- Determining the correct device classification
- Completing Foreign Manufacturer Registration (FMR)
- Appointing a reliable Japanese MAH/DMAH
- Preparing robust technical documentation and QMS compliance evidence
- Engaging with PMDA consultations early in development
A proactive regulatory strategy, strong local partnerships, and well-prepared submissions are essential for successfully accessing Japan’s lucrative medical device market.
References
- PMDA - Regulatory Body & Pathway:Regulations and Approval/Certification of Medical Devices | Pharmaceuticals and Medical Devices Agency
- MHLW - Registered Certification Bodies: Regulations and Approval/Certification of Medical Devices | Pharmaceuticals and Medical Devices Agency
- PMDA - JMDN Codes list:(https://www.std.pmda.go.jp/stdDB/index_en.html)
- PMDA - Class I Device Checklist : Medical Devices Required for Notification | Pharmaceuticals and Medical Devices Agency
- PMDA - Designated MAH: https://www.pmda.go.jp/english/about-pmda/0004.html
- PMDA - Required Documents (Foreign MFR): https://www.pmda.go.jp/english/about-pmda/0004.html
Share this blog
Read More Blogs

FDA QMSR Explained: Key Changes & ISO 13485 Alignment (2026)

US FDA Medical Device Registration Guide | 510(k), PMA & De Novo

Clinical Evaluation Plan (CEP) Guide for EU MDR & IVDR Compliance