

US FDA Medical Device Registration Guide | 510(k), PMA & De Novo

How to Register Your Medical Device in the U.S. FDA Market
The U.S. medical device market is one of the largest in the world, but it is also highly regulated. To ensure that patients receive safe and effective devices, the Food and Drug Administration (FDA) — through the Centre for Devices and Radiological Health (CDRH) — enforces strict controls before and after a device enters the market.
This guide explains how to navigate the U.S. FDA regulatory pathway, covering classification, submission routes, registration, labelling, timelines, and post-market obligations.
1. Confirm Whether Your Product is a Medical Device
The first step is to confirm if your product meets the FDA definition of a medical device under the FD&C Act. FDA provides guidance here: Is my product a medical device?.
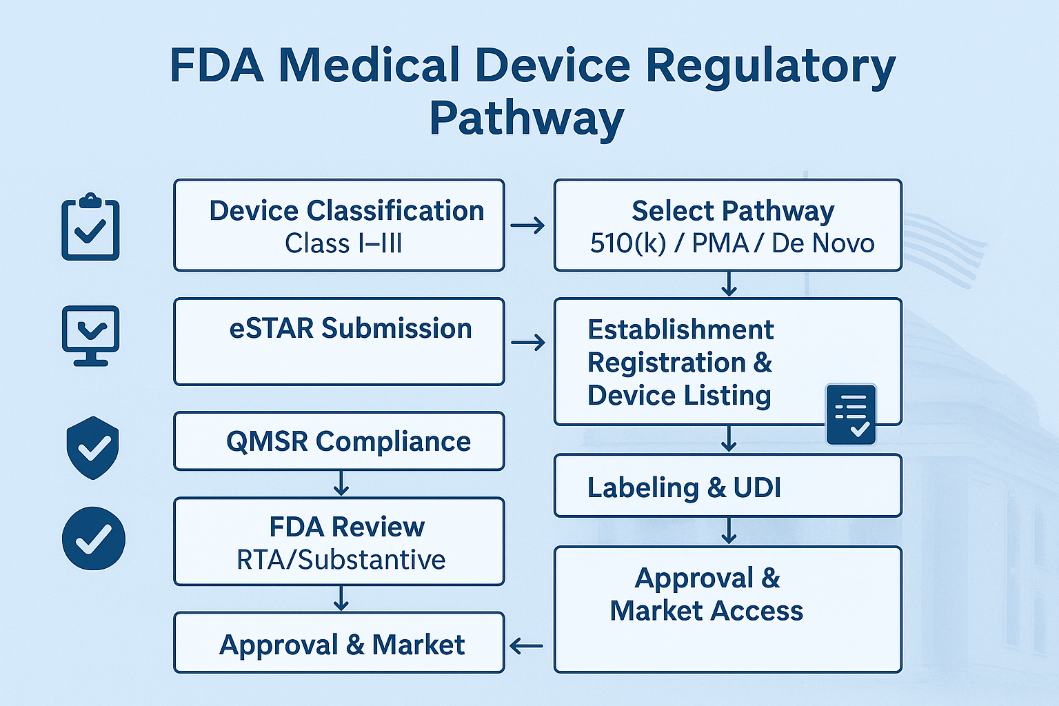
2. Device Classification
Devices are classified based on risk:
- Class I (Low Risk): General controls only. Some exempt from 510(k).
- Class II (Moderate Risk): General + Special controls. Usually require 510(k).
- Class III (High Risk): Require Premarket Approval (PMA) unless exempt.
Classification determines the type of submission and regulatory controls required.
3. FDA Marketing Pathways
- 510(k) Premarket Notification:
Most Class II and some Class I devices. Requires demonstration of substantial equivalence to a predicate device. Review time ~90 days. - Premarket Approval (PMA):
Required for most Class III devices. Involves clinical data and a rigorous FDA review. Review time ~180 days. - De Novo Classification:
For novel, low-to-moderate risk devices without a predicate. Successful De Novo creates a new classification. Review time ~150 days. - Other Pathways:
- Humanitarian Device Exemption (HDE): < 8,000 patients/year.
- Custom Device Exemption (CDE): For specific patients under physician order.
- Investigational Device Exemption (IDE): Allows use in clinical studies.
- Breakthrough Devices Program: Expedited review for novel, life-saving devices.
4. Submission via eSTAR
From October 1, 2023, all 510(k) submissions must be made electronically using FDA's eSTAR (Electronic Submission Template and Resource) unless exempt.
- eSTAR is a smart, interactive PDF template that ensures complete and structured submissions.
- Applications are submitted via FDA's Customer Collaboration Portal or Electronic Submissions Gateway (ESG).
5. Establishment Registration & Device Listing
- All establishments (domestic and foreign) must register annually with FDA via the FURLS/DRLM system.
- Devices must be listed, along with premarket submission numbers (if applicable).
- FY 2025 registration fee: $9,280; FY 2026 fee: $11,423.
- Foreign manufacturers must also appoint a U.S. Agent.
6. Quality Management System (QMS) Requirements
Manufacturers must comply with 21 CFR Part 820 Quality System Regulation (QSR). As of January 31, 2024, FDA updated this regulation to align closely with ISO 13485:2016, known as the Quality Management System Regulation (QMSR).
7. Labelling Requirements
Under 21 CFR Part 801 and UDI regulations (21 CFR Part 830):
- Device name, intended use, and manufacturer details.
- Warnings, precautions, contraindications.
- Expiry date, lot/serial number.
- Unique Device Identifier (UDI) on label and, where applicable, on device.
- Instructions for use and promotional material (considered part of labeling).
Non-compliant labelling is one of the most common reasons for FDA enforcement.
8. Medical Device Reporting (MDR)
FDA requires post-market vigilance under 21 CFR Part 803.
- Manufacturers: Must report deaths, serious injuries, and malfunctions.
- Importers: Must report deaths/serious injuries to FDA and manufacturer; malfunctions to the manufacturer.
- User Facilities: Must report deaths to FDA and manufacturer; serious injuries to the manufacturer.
Timelines:
- 30 calendar days: Standard MDR report.
- 5 working days: Urgent events needing immediate corrective action.
MDR helps FDA detect safety issues, initiate recalls, and protect patients.
9. FDA Review Timelines
- 510(k): RTA review (15 days), substantive review (~60 days), total ~90 days.
- De Novo: Typically, ~150 days.
- PMA: Filing review (45 days), scientific review + panel (if needed), total ~180 days (often longer for complex devices).
10. Conclusion
Registering a medical device with the FDA requires:
- Determining classification and correct pathway.
- Submitting via eSTAR (for 510(k)).
- Completing annual establishment registration and device listing.
- Complying with QMSR and labeling requirements.
- Maintaining post-market vigilance through MDR reporting.
By following these steps, manufacturers can ensure smooth entry into the U.S. market while meeting FDA’s high standards for safety, quality, and effectiveness.
11. References
- https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
- https://www.fda.gov/medical-devices/classify-your-medical-device/how-determine-if-your-product-medical-device
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/how-study-and-market-your-device
- https://www.fda.gov/medical-devices/device-registration-and-listing/how-register-and-list
- https://www.fda.gov/medical-devices/device-registration-and-listing/us-agents
- https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp
- https://www.fda.gov/industry/fda-user-fee-programs/medical-device-user-fee-amendments-mdufa-fees
Share this blog
Read More Blogs

Saudi Arabia Medical Device Approval | SFDA Regulations

Is DHF Compliance Risks Holding Back Your Medical Device Approval?

CDSCO Medical Device Registration in India – CDSCO SUGAM Portal Guide