

Clinical Evaluation Plan (CEP) Guide for EU MDR & IVDR Compliance

How to Develop a Comprehensive Clinical Evaluation Plan (CEP): A Complete Guide for Medical Devices and IVD Devices
Clinical evidence forms the backbone of medical device conformity under the EU MDR and EU IVDR. Whether for regulatory approval, CE marking, or lifecycle management, a Clinical Evaluation Plan (CEP) ensures that all clinical data—existing or newly generated—is collected and evaluated using a systematic, justified, and compliant process.
This blog provides a complete, structured approach to developing a robust CEP aligned with:
- EU MDR Annex XIV Part A Section 1(a)
- IVDR Annex XIII
- MDCG 2020-13 Clinical Evaluation Assessment Report (CEAR)
- MEDDEV 2.7/1 Rev. 4

Quick checklist (what the CEP must at minimum contain)
- Identification of GSPRs that require clinical data
- Intended purpose statement
- Target groups, indications & contraindications
- Intended clinical benefits and measurable outcome parameters
- Methods for qualitative & quantitative safety assessment (residual risks, side effects)
- Parameters for benefit–risk acceptability (state of the art)
- How special components (pharma, animal/human tissue, etc.) are handled
- Clinical development plan
- Document metadata: reference, version, date, author, approver
Step 1 — Prepare the CEP skeleton
Create the controlled document and including Document title, Manufacturer reference, Version/Rev No., Date of issue, Author(s), Approver(s), Next review date & change history
Why: MDCG 2020-13 expects CEP metadata so reviewers can trace versions and approvals.
Step 2 — Define intended purpose & scope
Write a precise intended purpose covering:
- Medical indication(s) / clinical condition(s)
- Therapeutic or diagnostic function (mechanism of action)
- Target patient population (age, comorbidities)
- User type (HCP / lay / home use)
- Clinical setting (hospital, outpatient, POCT)
- Device variants included in the evaluation
Why: Every claim, GSPR mapping and study design flows from the intended purpose.
Step 3 — Map GSPRs to clinical evidence needs
Create a GSPR matrix:
- List Annex I GSPRs (and any device-specific GSPRs)
- For each GSPR: note whether evidence is non-clinical, clinical, or both
- Identify which GSPRs require clinical data (safety/performance/use-error, clinical benefit claims)
Deliverable: GSPR → Evidence mapping table (keeps your CER auditable).
Step 4 — Define clinical claims, clinical benefits & outcome parameters
For each clinical claim:
- State measurable clinical outcome(s) (primary and secondary endpoints)
- Define acceptance criteria (numeric thresholds, timeframes)
- Specify safety endpoints and residual risk indicators
Example:
Claim: “Reduces time-to-diagnosis for X” → Outcome: mean time (minutes), % correct diagnoses, clinical impact on treatment decision.
Why: Clear endpoints determine literature selection, study design and PMCF metrics.
Step 5 — Specify target groups, indications and contra-indications
Document:
- Included populations (age ranges, disease stage/severity)
- Excluded groups and rationale (contraindications)
- Subgroups requiring special evidence (e.g., paediatrics, immunocompromised)
Why: Notified Bodies expect explicit population boundaries and justification.
Step 6 — State-of-the-Art (SOTA) & benefit-risk acceptability parameters
Outline SOTA:
- Clinical guidelines, standard of care, alternative therapies/devices
- Benchmarks for acceptable benefit-risk (sensitivity/specificity, complication rates, QoL metrics)
- Parameters you will use to judge acceptability for each indication
Deliverable: SOTA summary with references and target performance thresholds.
Step 7 — Plan your literature & data search (systematic review strategy)
Document a reproducible search plan:
- Databases: PubMed, Google Scholar, Cochrane, regulatory databases
- Search strings and date range
- Inclusion/exclusion criteria (device vs. similar device, population, study design, language)
- Screening process (titles → abstracts → full text)
- Data extraction fields (study design, population, endpoints, results, bias)
Why: EU MDR/ IVDR Annex XIV(b) requires identification of available clinical data via systematic review.
Step 8 — Appraisal methodology (how you will judge evidence quality)
Choose appraisal tools and explain use:
- Risk of Bias and Grade the Strength of Evidence — evaluating how reliable each study is and how strongly it supports the device’s safety and performance.
- Device-Specific Appraisal Criteria — considering factors such as user training requirements, relevance of the chosen comparator, and whether the data represents the current generation of the device.
Step 9 — Equivalence strategy (if you plan to rely on equivalent device data)
If claiming equivalence, document and justify:
- Technical comparison table (design, materials, software, energy, etc.)
- Biological comparison (materials in contact, contact duration, leachable)
- Clinical comparison (indication, population, performance metrics)
Evidence access statement (confirm you have sufficient access to competitor technical/clinical data)
Red flag: If you cannot access competitor technical data, equivalence is weak — plan confirmatory data instead.
Step 10 — Identify evidence gaps & define required new data
Using the appraisal, list gaps:
- Missing endpoints, insufficient population representation, poor-quality studies
- For each gap: choose action — use PMCF, run a feasibility/pivotal study, collect registry data, or perform a targeted PMCF study
Deliverable: Evidence gap register with assigned owners and timelines.
Step 11 — Clinical Development Plan (CDP)
The CDP outlines the staged progression of clinical activities as required by EU MDR Annex XIV—from exploratory studies (bench, usability, early feasibility) to confirmatory pivotal investigations and finally PMCF. Each stage includes predefined milestones and acceptance criteria (e.g., feasibility safety thresholds, pivotal study endpoint success). This structured approach ensures the device demonstrates safety, performance, and a favourable benefit-risk profile before market approval and continuously after the market placement.
Step 12 — Safety assessment methods (qualitative & quantitative)
Define how you will measure safety:
- Qualitative: complaint review, usability findings, case reports, HF analyses
- Quantitative: Adverse event rates per device-exposure, incidence per 1000 uses
Link to risk management (ISO 14971): residual risks and evaluation thresholds
Step 13 — PMCF / PMPF (Post-Market Clinical/Performance Follow-up) plan details
Specify:
- PMCF/PMPF objectives (confirm performance, detect rare AEs, long-term outcomes)
- Study designs (prospective registry, cohort, retrospective data mining)
- Sample size rationale, endpoints, follow-up duration, data collection tools
- Triggers for escalation (e.g., AE rate above threshold) and acceptance criteria
Why: PMCF is required unless justified otherwise — and is mandatory for many high-risk devices.
Step 14 — Roles, responsibilities & governance
Define:
- CEP owner and authorship chain
- Multidisciplinary review team (Clinical, RA, QA, R&D, Biostatistics)
- Approval process & version control
- Frequency of review (e.g., annually or sooner if new data emerges)
Deliverable: Responsibility matrix.
Step 15 — Integration with other QMS elements
Ensure links to:
- Risk Management File (RMF) - trace residual risks → clinical evidence
- PMS system - use vigilance and complaint data as clinical evidence sources
- Technical Documentation - place CER and CEP in TDF
- Labelling & IFU - ensure consistency with intended purpose and claims
Why: Regulators check traceability across documents.
Step 16 — Reporting & documentation standards (MDCG 2020-13 alignment)
Final outputs: CEP, CER (or PER for IVDs), PMCF plan and updates
Specific Requirements Under MDR vs IVDR
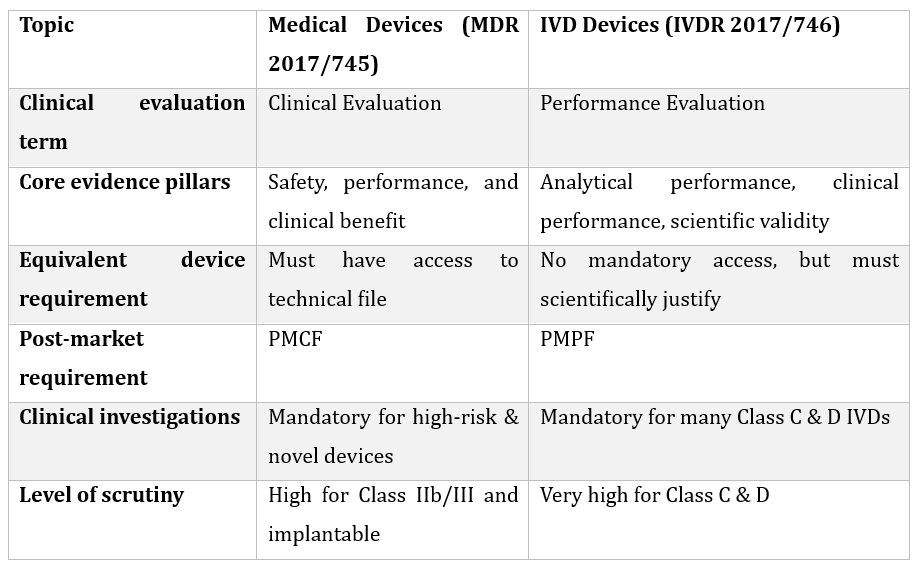
Conclusion
A well-designed CEP is critical for ensuring EU MDR/IVDR compliance and demonstrating that your device is safe, effective, and supported by solid scientific and clinical evidence. By following this structured approach—aligned with EU MDR/IVDR Annex XIV, Annex XIII, MEDDEV 2.7/1 Rev.4, and MDCG 2020-13—manufacturers can produce a robust, audit-ready CEP that withstands scrutiny from Notified Bodies.
Share this blog
Read More Blogs

US FDA Medical Device Registration Guide | 510(k), PMA & De Novo

FDA QMSR Explained: Key Changes & ISO 13485 Alignment (2026)

Unique Device Identification Made Simple with V-Reg Solutions